Contents:
A PCA, probably the most basic analysis we are doing. The script merge_and_pca.sh merges the genotypes of a comparative data set with the information from your sample. If there are reads covering a respective SNP position, one read is randomly drawn from them if that carries one of the known SNP alleles (and no T or A at a transition site) and that allele is assumed to be homozygous in our ancient individual. Later all modern individuals are also made homozygous by randomly picking one allele per SNP.
Since you usually don’t want to make a PCA with all worldwide populations, you shoud specify a file naming all modern populations for your PCA. Please check the .tfam file corresponding to your tped to find out what populations are available in the data set, for some datasets there are a file ending with -allpop.txt which lists all populations in the dataset. You also need to provide the correct for your SNP data (hs37d5.fa (hg19, needed for Human Origins) or human_b36_male_nohaps.fa(hg18, needed for HGDP et al.)). Please provide the full path to all of these files.
sbatch -A $proj -p core -n 2 -t 2-0:00:00 -J PCA-${sample} merge_and_pca.sh ${BAMFILE} ${TPEDFILE} ${POPLIST} ${REF} ${TRANS} ${PMD}
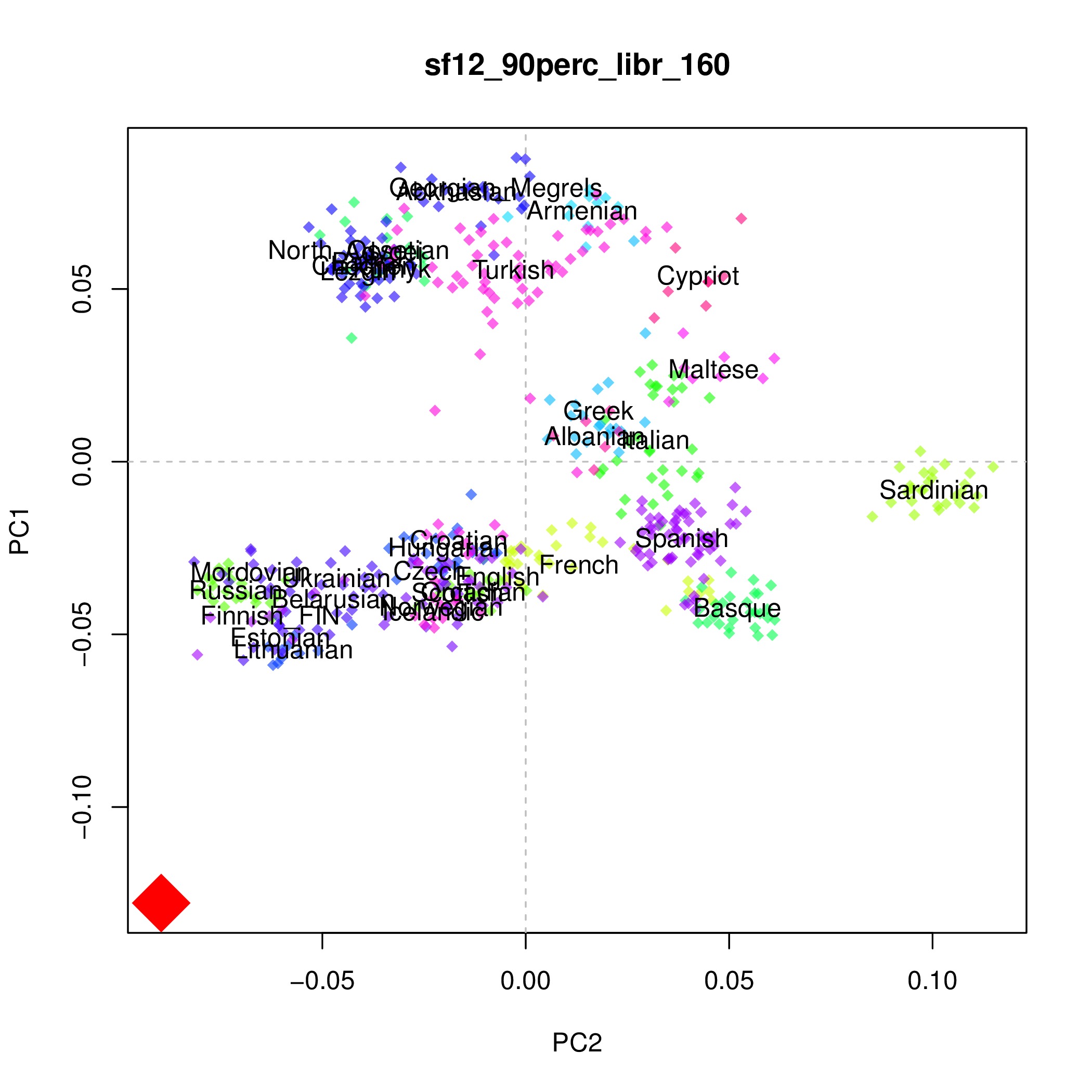
##TRANS - 0 or 1 (default 0) if 1, filter all transition sites. if 0, only filter transition sites if the ancient individual shows a T or AAfter a couple of hours you will have a pdf file (together with many other files) showing your sample as a big red diamond together with all modern populations. This is a PCA with our favorite high coverage sample SF12:
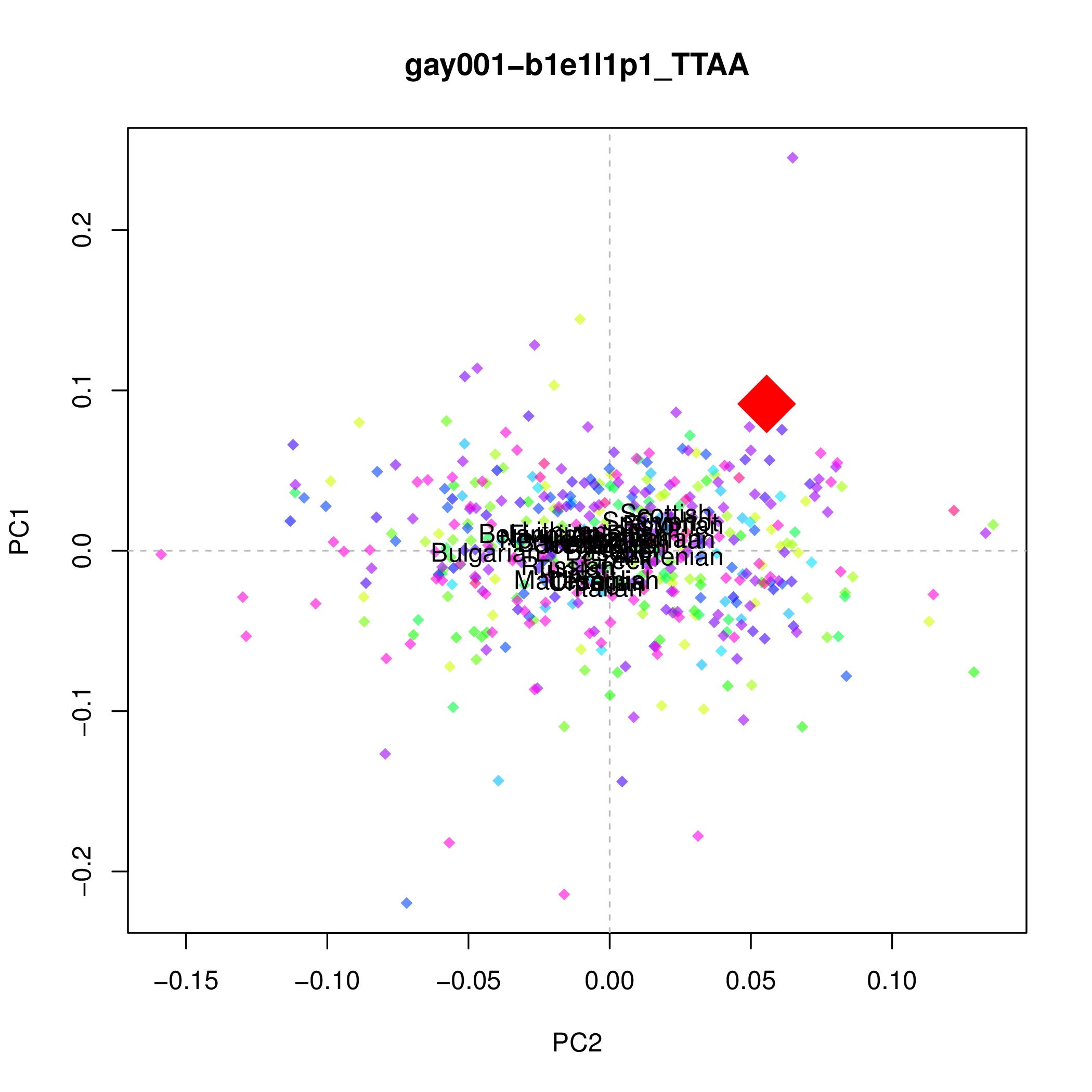
To check how many SNPs have been used to generate your PCA, you count the number of rows there is in the mapfile, wc -l *.map. If you have a low coverage sample there will not be enough SNPs to properly distinguish the modern populations from eachother. Your PCA will look something like this instead:
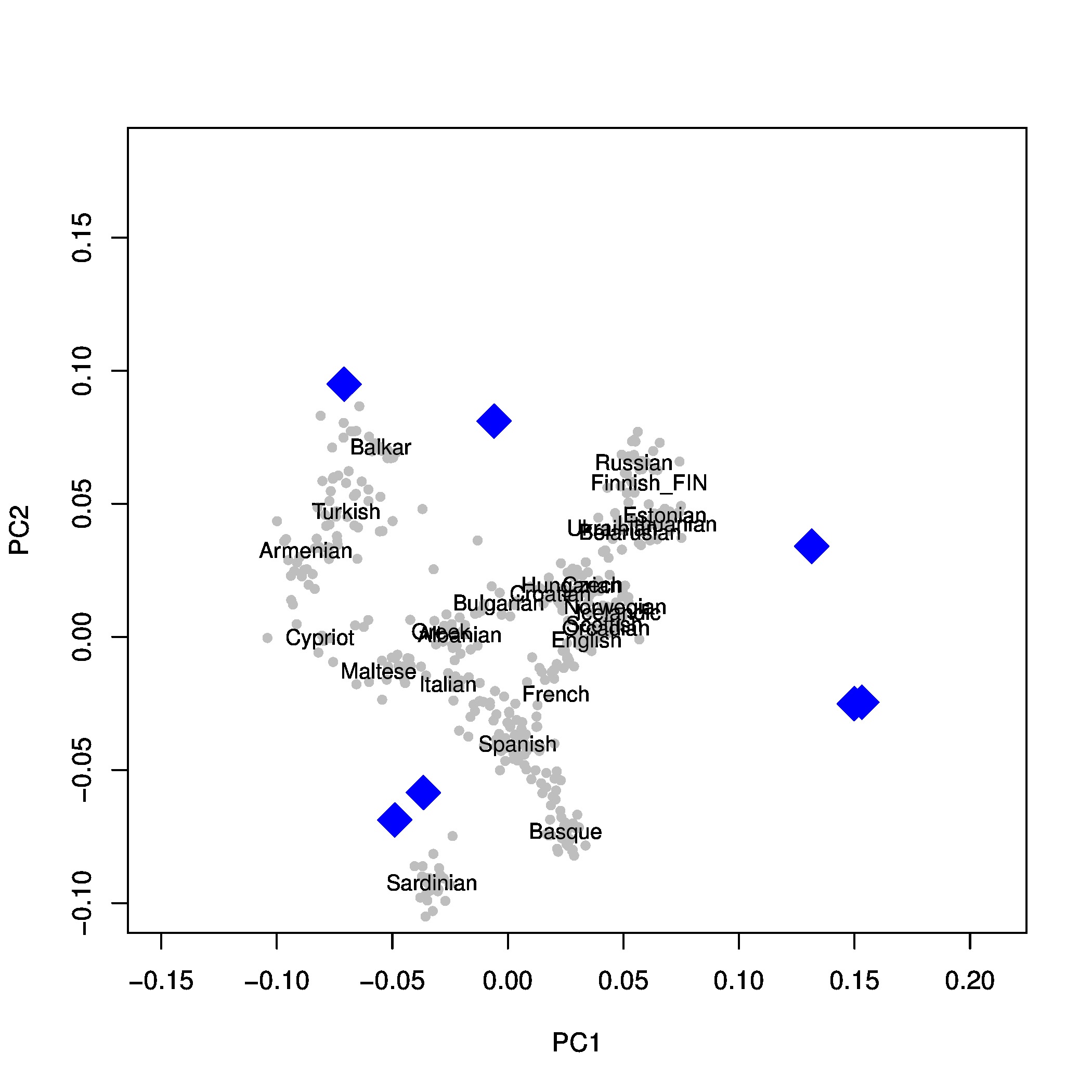
We are using Procrustes transformation since Skoglund et al. (2012) [1] to display multiple ancient individuals in the same PCA. It is one of the possible techniques to cope with all the missing data in the different samples.
You need to do a PCA for each of your ancient samples (keep the .tfam and .evec files) and one for the reference SNP data (but just the subset of the populations you are using, you can use plink --keep to select a subset of a tped file). Since different projects require different ways to display the data,
we provide only an example script in the central directory (procustes_example.R). Please make a copy of this script in your local directory and then modify it. You’ll have to modify the names of the .tfam and .evec file of you reference PCA in the first lines.
Then you can run the script as Rscript procustes_example.R .evec .evec .evec etc. (just give a list of all your individual .evec files as command line arguments). Don’t forget to module load R/3.4.0 R_packages/3.4.0 before running procustes. You might want to change colors, symbols and add a legend, please modify the code of your local copy accordingly.
We have started to move away from Procrustes transformation since there is a risk that a single SNP could be driving the entire position in procrustes. Instead we are using lsq projection more and more which has two main advantages: ancient samples are only projected on the modern PCs (no risk of them driving any PCs due to aDNA related technical issues or only few SNPs covered) and they can be run in one go for all samples in a tped/tfam file (which can be the same file you use in other types of analysis, e.g. ADMIXTURE or f statistics). (We still need to explain how to generate such a file.)
This is a description for how to use a script for running smartpca with the options lsqproj and shrinkmode. The script pca_lsqproj.sh assumes that you have a merged set of tped/tfam files containing data for both ancient and modern populations. You should also have a file listing the modern reference populations you want to use (one per line) and a file listing the ancient populations you have in your tped/tfam files that you want to project. Note that the Populations IDs in <modern reference pops> must exactly match the IDs in the tfam, while population IDs in <ancient pops to include> can be more general (e.g. HG_SE instead of HG_SE_SF12) – No error messages when this is violated but weird results may occur!!
sbatch -A $proj -p core -n 2 -t 2-0:00:00 -J PCA pca_lsqproj.sh <name of tped/tfam file> <modern reference pops> <ancient pops to include>
After a couple of hours of running, the output will be a .evec and a .eval file. You can plot the information from .eval using e.g. GNU R.
Bibliography
| [1] | Pontus Skoglund, Helena Malmström, Maanasa Raghavan, Jan Storå, Per Hall, Eske Willerslev, M. Thomas P. Gilbert, Anders Götherström, and Mattias Jakobsson. Origins and Genetic Legacy of Neolithic Farmers and Hunter-Gatherers in Europe. Science, 336(6080):466–469, April 2012. URL: http://science.sciencemag.org/content/336/6080/466, doi:10.1126/science.1216304. |